

Rare diseases can help illuminate treatment opportunities in more prevalent diseases but often lack the financial support and public awareness to do so.
The National Institutes of Health defines a rare disease as one that affects fewer than 200,000 people in the United States at a given time. There are nearly 7,000 rare diseases in the U.S., and nearly 30 million Americans are living with one.
Since 2008, international research and education organizations have raised awareness at the annual Rare Disease Day. The University of Minnesota participated Friday by sponsoring events that highlighted rare disease research efforts by the Center for Orphan Drug Research and the Stem Cell Institute.
“It’s a rare disease day, but I can tell you from personal experience that if this is you, or if this is your parent or your kid, it’s not rare anymore,” said Stem Cell Institute Director Dr. Jakub Tolar. “They don’t think, ‘Oh, I have a rare disease.’ They think, ‘I have a disease.’”
Unexpected innovations
Funding for and education about rare diseases pales in comparison to that of better-known diseases, said Center for Orphan Drug Research Endowed Chair James Cloyd.
It is sometimes challenging to recruit more young professionals in the study of rare diseases, he said. He counts himself lucky to have about half a dozen
undergraduate students work on his research team.
“We are not only putting together teams that can bring some new knowledge, but we also are very heavily focused on mentoring and educating the people that will come after us to take this even further,” Tolar said.
It’s important to study rare diseases, he said, because they’re often the pathway to learning more about common diseases — which are often more complex. The majority of rare diseases are genetically linked and occur in childhood, he said, making them less likely to be altered by outside factors and therefore easier to study.
University research on the rare disease fanconi anemia, for example, contributed to the discovery that genes involved in it also make people more susceptible to breast cancer.
Findings like these have increased over the last few decades.
Lawrence Weaver, former dean of the College of Pharmacy, helped author some amendments to the 1983 federal Orphan Drug Act, which stimulated nationwide research of orphan drugs — medications that often haven’t been fully researched for their possibilities in treating rare diseases because of the higher demand for treatment of more common diseases.
Since then, the U.S. Food and Drug Administration has approved more than 400 drugs and other medicinal products for development for rare diseases, compared to fewer than 10 in the decade before the act passed.
Targeted questions, serendipitous successes
Cloyd admitted that in rare disease research, many beginnings aren’t formulaic.
But he said it helps to be surrounded by smart people with smart questions who want to use the University to collaborate in solving problems.
The Center for Orphan Drug Research repurposes existing drugs by combining the efforts of medical experts and students across the University. Cloyd said faculty members often come to the center with a problem an existing medication has, or with an overall lack of medication, and ask the center to investigate it.
One such question, and the center’s resulting research, has led to hope for better treatment for boys with the rare disease adrenoleukodystrophy, or ALD.
The disease is often hard to spot in young boys because its symptoms typically don’t show for years. Eventually, a boy with ALD develops signs such as lack of attention, slow loss of hearing or sight, or inexplicably tan skin. But as the disease progresses, it causes irreversible loss of brain connections from oxidative damage and inflammation, which can result in serious seizures. Boys at this late stage almost always die, unless they receive a bone marrow transplant in time.
Five years ago, prompted by a faculty member’s curiosity, the center began research on a drug that was a known antioxidant. The center explored how it might be able to combat oxidation in the brains of boys with ALD, and it found that the drug was capable of improving survival in the late stages of the disease.
“If I can make [people] feel how tragic this is, how important this is; if I can for a millisecond make them feel this is your brother, this is your son; then I have accomplished something,” Tolar said.
‘Not finished yet’
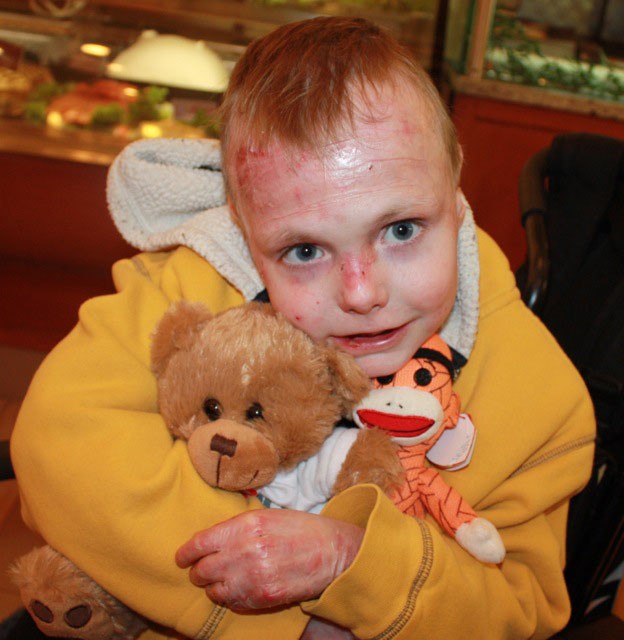
Charlie Knuth’s epidermolysis bullosa makes his skin so fragile that it could tear or blister with even a slight nudge. Out of a million newborns, less than one has this disease.
Before his stem cell transplant in 2010, bandages covered about 90 percent of Knuth’s body all the time, said his mother, Trisha Knuth. Baths and dressing changes could take anywhere from five hours to a full day.
Now, his wounds cover only about 5 percent of his body, and his dressing changes take an hour or less.
Less than a decade ago, Trisha Knuth described access to information about her son’s rare disorder as inadequate. Since then, there have been huge strides, she said — but there is more work to do.
“Yeah, his life is better, but no, we’re not finished yet, and it’s still very hard,” she said. “I’m looking forward to the future of research and translational medicine.”